From Muck to Mauve: the creation of modern dyes from coal tar

📖 40 min read • Aniline and the birth of the German chemical industry
Photo credit: James St. John. Flickr [CC BY 2.0]
Content notice: this post discusses the early dye companies of industrialised pre-WWI Germany that were at one point put to horrific wartime use. World War II is alluded to in a literary work that was famously denied a Pulitzer prize in part because of its confronting content.
In this post I’m aiming to give a broad-brush overview of how the near-infinite array of colours we see in textiles today were wrought from some of the most murky carbon substances known. This post will focus on purple (specifically, mauveine) but will meander through the history and chemistry of the blue hues.
I’d like to write more on specific dyes in the future because they have so many fascinating chemical properties. For example, indigo forms from a dimer using the indole group common in biochemistry, placing it within striking distance of tryptophan (amino acid \(W\)) in the biosynthetic pathway.
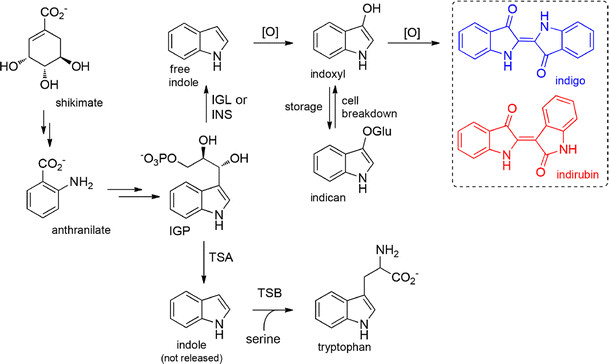
Indigo is made from joining two slightly modified indole molecules. Indole is such a common structure in biology that it is used to make tryptophan, one of the 20 standard amino acids, which can reveal protein folding properties through fluorescence. —Synthesis diagram from Plant Cell Reports, 2016, 35, 2449–2459.
The development of dyes used to be covered in a (rarely taught) NSW syllabus option, “Chemistry of Art”, that touched on everything from indigenous ochres to transition metal pigments and atomic spectroscopy, but is sadly now gone.
For those interested in colour more generally, I can recommend “The Secret Lives of Colour”. Its chapters are arranged by colour, making it addictively easy to pick up and dive into each hue. I also recommend “The History of Colour” for an academic history of the conception of colour — Chapter 2 starts with synthetic purple and details the explosion in commercially valuable tints that followed. If the development of industrial dyes interests you, BASF have an account of their history on their website including the “Aniline Women” who worked there.
Some correspondence by scientists picturing this new cyclical chemical reality is contained in “Image and Reality: Kekulé, Kopp, and the Scientific Imagination” , providing primary sources around the murky question of whether he actually dreamed the snake. Image and Reality gives interesting rapid recount into the depictional development of atomic theory, including the radically clear-eyed early experimental proof by Alexander Williamson who provided, using etherification reactions, his pictorial theory of “an actual image of what we rationally suppose to be the arrangement of constituent atoms in a compound”.
Williamson’s visual foresight gave a hazy glimpse at perceiving how constant molecular motion created reactivity, and many other scientists saw farther than the peers of their day by picturing the non-empirical.
Pauvre mauve 🪻
There’s an abundance of green in the natural world (likely because plant chlorophyll is tuned for reliable photosynthesis), but blues and purples stick out because they are so rare in nature. Maybe that’s why blue is humanity’s favourite colour. The rare examples of natural blue are unusual — bird and butterfly wings have to resort to optical trickery (metamaterials); blueberries get their name and antioxidant health craze because their skin is packed with the crop pigment anthocyanin; the blue warning signal of the poison dart frog doesn’t arise from its toxin (pumiliotoxin), but comes from skin iridophore cells containing stacks of reflective guanine platelets; and lobsters change spectacularly from blue to red upon boiling due to heat denaturing their crustacyanin.
Almost all natural pigments in the blue end of the spectrum (and others) are pigments built from the anthocyanidin skeleton shown above. —Wikimedia Commons, NEUROtiker, 5 August 2008.
{kind=link}
Similarly, there’s an almost complete absence of natural purple. Purple cabbage and eggplant contain the same anthocyanins as blueberries, similarly for violet’s flowers whose colour may be striking but is not colourfast when used to dye fabric. So, when nature was our only source of chemicals, people’s desire to wear the spectrum of ‘cool’ colours was met by crushing the leaves of the somewhat drab woad, and the perennially popular indigo — the name given to both the plant and one of Newton’s seven ‘ROYGBIV’ colours partitioning the rainbow.
The only concentrated, colourfast sources of organic purple come from the seas. Aplysia sea slugs spray purple aplysioviolin molecules when threatened. Sea hares build this ink up in their glands by eating red algae; stripping the colourant molecule from the algae’s light harvesting proteins and concentrating an aplysioviolin derivative. A painstaking, but more reliable, way to obtain natural purple is to extract ink glands from the Murex sea snail. The ‘Tyrian purple’ dye harvested from these snail glands was, and is, outrageously expensive, fetching around $3,000 USD/g in the 21st century. Tyrian purple is in fact a brominated derivative of indigo, synthesised using a bromoperoxidase enzyme unique to Murex. Scientists still have not confirmed the active metal within the Murex’s bromoperoxidase enzyme (most bromoperoxidases use the rare vanadium), or which gene in the sea snail encodes for this purple producing enzyme.
Lapis lazuli and other blues of value 🔵
For making paints and dyes, even the colour diversity found within the minerals of the Earth was of little help; the naturally occurring mineral pigments are prone to dull hues, rapid fading, and discolouration into green (e.g. azurite). Synthetic mineral blue dyes were created in ancient times from copper silicates (mixed with calcium - “Egyptian Blue”, or barium - “Han Blue & Purple”), or otherwise just from indigo ‘baked into’ a mineral substrate, as detailed in a Chemical Society Reviews article on “The invention of blue and purple pigments in ancient times”. However, Han Purple did not provide a pure purple, its appearance was due to red copper oxide impurities. Similarly, pure, brilliant blue mineral pigment was only provided by eye-wateringly expensive lapis lazuli (ultramarine), mined only around the mountains on the North-Eastern tip of Afghanistan. Ultramarine would cost up to 100x other pigments, but its dazzling effect was valued in religious iconography across Asia and Europe, and has notable use in famous paintings (e.g. the blue headscarf in Girl with a Pearl Earring). Since mineral pigments and synthetics paled in comparison to “true” ultramarine, lapis lazuli was in demand well into the 19th century. The physical origin of the intense blue appearance of ultramarine is chemically interesting: rather than a metal at the centre of the mineral, the colour comes instead from a negatively charged trisulfur radical (S\(_3^{\bullet-}\)).
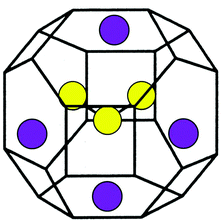 The negatively charged trisulfur radical caged in a zeolite with its surrounding sodium ions. —Chemical Society Reviews, 2013, 42, 5996-6005 adapted from Inorganic Chemistry 2002, 41, 11, 2848–2854.
The negatively charged trisulfur radical caged in a zeolite with its surrounding sodium ions. —Chemical Society Reviews, 2013, 42, 5996-6005 adapted from Inorganic Chemistry 2002, 41, 11, 2848–2854.
Now with modern chemistry, blue hues can be created that are so striking that an artist can make a multi-million dollar name for themselves with one famous blue dye, and scientific careers and reputations can be built upon chemists’ prowess to manufacture the bluest blue. The chemistry to produce commercially abundant purple dyes was however only cracked in the 19th century, and with it opened up new possibilities in organic synthesis. Even now, there is unplumbed depths to these cool hues: the colour terminology for purple and violet is murky, our visual perception strains near the UV wavelengths, and even the colour range of our computer monitors come up short.
Porphyrogeniture 👑
Purple has long signified royalty and power in the ancient world, to the point that its use was restricted to high office; not just due to the expense of natural purple dye, but often enforced by social codes and law. Famously, Roman officials would colour their formal attire (togas) with a strip of purple along the border. This style, the toga praetexta (from Praetor, a magistrate position below Consul) wasn’t solely restricted to appointed officials, but likely had a religious origin and was recorded as being worn by priests and pre-adolescent children of fortunate birth. However, a toga fully clad in purple became restricted to Roman consuls and generals performing a Triumph, and later to the Emperor alone. Nero limited the use of purple to the imperial court, and the unsanctioned wearing of purple was even codified as an offence punishable by death by Theodosius II.
The association of the purple hue with royalty extended beyond clothing, and Byzantine (Eastern Roman) queens would give birth to future rulers in rooms decorated with a deep-purple Imperial Porphyry stone (from Ancient Greek porphyra — purple). This birthright to distinction through colour was enforced from cradle to grave, as Emperors were buried in tombs made from porphyry, some survive today outside the Archaeological Museum in Istanbul. The purple name echoes throughout history, such as the sisters Zoe and Theodora Porphyrogenita who co-ruled the Byzantine Empire after a popular revolt.
{kind=link}
{kind=link}
Born in the purple 🟣
Ironically, despite the incredible rarity of steadfast purple in nature, purple was the first in a tidal wave of synthetic industrial dyes, bringing the colour to the masses and, as it goes with fads, becoming so commonplace as to be unfashionable quickly after reaching the mass market. The discovery of artificial purple dye (mauvine) was entirely accidental, but the chemical knowledge to synthesise it at massive scale required unravelling some of the fundamental mysteries in organic (carbon-based) chemistry; in the process clarifying how electrons create molecular structure and govern the interactions of dye molecules with light.
The first artificial purple dye was synthesised by William Henry Perkin in 1856 from an accidental side discovery. Perkin’s motivation was not encolouration, but empire. What Perkin was searching for was a cheap synthetic route to quinine, an antimalarial substance isolated from cinchona tree bark, useful for stationing soldiers in colonies of the British Empire. Perkin’s synthesis attempts were misguided from the start, having only the empirical formula of quinine (C\(_{20}\)H\(_{24}\)N\(_{2}\)O\(_{2}\)) and no idea of its structure, he explored combining two equivalents of an organic starting material whose masses would sum to the total atom count of quinine once oxygenated. In reality quinine’s structure is quite complex and the quest to irrefutably create it from scratch kicked off 150 years of investigation and controversy into its difficult total synthesis. Incidentally, quinine is the source of tonic water’s ultraviolet glow under blacklight.
{kind=link}
What Perkin saw in his experiment was not emissions de-energised from the ultraviolet, but instead found that when diluting his failed synthesis product with alcohol during wash-up the solution turned purple. He opened up a dyeworks in Greenford the next year. The success of a synthetic dyes would lead to a complete collapse of the economic harvesting of natural dyes such as madder root.

Perkin’s mauve was a runaway success but through its overuse in fashion it came to be see as tawdry. The use of the gaudy colour fell out of favour for everyday wear. —Journal of the Society of Dyers and Colourists, Nov. 1906, via the Science History Institute .
However, Perkin’s happy discovery at 18 years old started from being encouraged to experiment with the idea of synthesising quinine by August Wilhelm von Hofmann while working as his assistant. Perkin continued his experiments out of the lab, and in his own shed, in order to trial creating proper dyes and keep the profits. The raw organic feedstock to make these dyes became available at a commercial scale due to breakthroughs in organic synthesis and industrial processes to make flammable gas from coal tar. Though able to unlock amazing synthesis routes, Perkin, Hofmann and chemists were working on organic materials without knowing their true atomic connectivity. The substances they worked with were termed “aromatic” compounds due to a few representatives having a strong smell (e.g. benzoin, in incense), but the molecular structure of these aromatics was a puzzle from only their empirical formulae and reactivities.
Muckraking ⚗️
Coal tar’s shimmering opacity hides its chemical complexity. In the murky pitch black mixture are thousands of different hydrocarbon species, and until modern petroleum extraction they were the major feedstock to synthesise organic molecules. The stodgy goop’s appearance obscures a mosaic of complex carbon skeletons, ripe to transform into a rainbow of complex organics furnished from chemical reactions. August Wilhelm von Hofmann was one of the pioneers in these experiments, considered by science historians to be one of the “fathers of synthetic chemistry”. He and Jamaican born chemist John Blyth were the first to use the term “synthesis” in a publication.
Hofmann’s interest in coal tar can be seen by the title of his first publication in 1843 “Chemische Untersuchung der organischen Basen im Steinkohlen-Theeroel” (“Chemical Investigation of the Organic Bases in Coal-Tar Oil”). Hofmann was able to isolate both aniline (the basis for dyes — mauvine was also called “aniline purple”) and quinoline (a degradation product of the quinine — Perkin was also interested in finding) from coal tar.
Experiments with coal tar and other organic synthesis projects were a productive foundation for Hofmann to build a research career on. The structure of Hofmann’s lab is seen as the precusor to the modern industrial chemistry research lab and was a source of many discoveries, such as the isolation of benzene (then benzole) by his British student Charles Mansfield. Additionally, Hofmann was the first to build physical ball-and-stick “glyptic formulae” models to display an object to embody the atomic connections inside molecules. His croquet ball colour choices of black for carbon, white for hydrogen, and red for oxygen remain in wide use today.
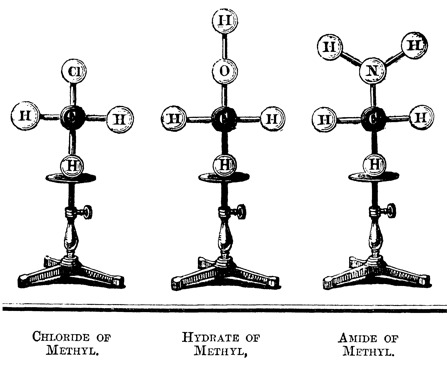
August Wilhelm Hofmann, “On the combining power of atoms”, Proceedings of the Royal Institution of Great Britain, 1865, 4, 401–430
Despite Hofmann’s lab being in Imperial College London (he moved to the University of Berlin in 1865), and Perkin being the first to commercialise aniline dyes in Britain, this nascent dye and chemical industry was to come to be dominated by Germany, largely due to economic and technological factors. A key factor was the industrial production of synthetic liquid and gaseous fuels from coal tar, invented in Germany in 1920s. As Germany had vast coal deposits and almost no natural petroleum reserves, this Fischer-Tropsch synthesis of fuel from coal tar was crucial for the energy, transport, and military viability of Germany. By making huge quantities of syngas from coal tar deposits and building industrial chemical plants, the feedstocks and techniques for dye creation were linked to plentiful industrial production in the Rhineland.
The economic advantage from industrial scale organic synthesis far outgrew the impact of only dyestuffs. A letter to Nature in 1934 puts it plainly:
“WHILST the original discovery of a coal-tar dye was made by an Englishman… The consequent decline of the British coal-tar colour industry was already well marked in 1875, and in 1886 had proceeded so far that 90 per cent of the dyes then used in Britain were of foreign manufacture… It is not an overstatement to say that the development of this highly scientific and extremely profitable industry in Germany instead of in Great Britain had enormous, if not decisive, political and economic effects both before and during the War.”
Enclosed with this submission to Nature is an additional letter where John Brunner (co-founder of a chemical firm that would later merge to become Imperial Chemical Industries) opines in 1915 that, if he and his chemist brother had money in their ‘teens’, their fascination with aniline dyed silk skeins would have driven them to keep the coal tar chemical industry in Britain.
Coal tarred and feathered 🪨
The importance of the profits from dyes in the founding of chemical companies can be seen in the names of industrial giants which are still around today, for example: BASF - Badische Anilin- und Sodafabrik (Baden Aniline and Sodium Carbonate Factory). BASF remains the largest producer of chemicals in the world: a multinational across 80+ countries with an annual revenue around €70 billion. Its history has touched on many chemical process famous in school curricula: BASF’s main trade was in dyestuffs until the 1900s; the rise of this company was meteoric after Haber and Bosch invented their method to turn nitrogen from the air into fertiliser; BASF invented polystyrene in the 1930s; and the sodafabrik part of BASF is infamous to New South Wales high school students who had to memorise the conditions of the Solvay process as part of their Industrial Chemistry module.
Under Nazi rule before and during WWII, BASF would be united with other industrial titans into the chemical conglomerate I.G. Farben (loosely, “The Colour Cartel”) of notorious and horrific historical fame. This conglomerate was founded by Carl Bosch through merging BASF with Hoechst (now merged under Sanofi), Bayer (widely known for aspirin), and a few other smaller chemical companies. After WWII, IG Farben was seized by Allied forces — its slave and exterminative labour operations inside concentration camps across Europe were shut down; many executives responsible for this deliberate and callous mass cruelty, industrially exploiting human lives, received light sentences at the Nuremberg trials that did not match the consequences of their crimes. The IG Farben conglomerate split up into its constituent components. In the reconstruction period, however, several of these companies such as BASF were refounded and allowed to rehire many of the same directorial staff who had served at the behest of depraved, horrific warmongers. These challenges to societal and base human conscience, the reshaping of entire industries, the discovery of new medical compounds (antifungals and antibiotics), and unfurling the spectrum of dyes for fabrics of all colours, resulted from an insight into the puzzling carbon skeleton structure of these newly synthesisable “aromatic” substances.
Benzene: The daydream snake 🐍♻️♾️
Benzene is a molecule, whose empirical formula defied the trends determined from other hydrocarbons. It took 40 years from the experimental isolation of benzene to solve its chemical structure. It clearly had a lower H:C ratio than “saturated” hydrocarbons, yet did not easily accept addition of new atoms like unsaturated hydrocarbons, and its boiling point just didn’t fit with other organics that matched its mass. This puzzled chemists even after they nailed down the true atomic weights of elements and were sure of the empirical composition of molecules. Any chemistry student will tell you it’s a hexagon. Benzene-as-a-hexagon is so commonplace to modern chemists that you can even buy hexagon stencils to help you breeze through your organic chemistry homework. But how was this shape arrived at?
The story of the solution to puzzling structure of C\(_{6}\)H\(_{6}\) is one of the few times instructors can retell a founding myth; like Romulus and Remus being raised by wolves then raising Rome, or Mendeleev playing atomic solitaire until the cards of the Periodic Table just fell into place. (Though I’m favourable to alternative depictions, or spiralling out once in a while). Chemistry teachers get to share this dramatic origin story — a scientist called August Kekulé simply dreamed of benzene and woke up with the solution!
In more detail, while dozing off on the last bus home from a friend’s place in London, Kekulé visualised chains of atoms in a whirling dance (at that time chemists were still figuring out how to apply recent studies by physicists on particle motion to their empirical chemical formulae). Suddenly, one chain of atoms seizes its own end, much like a snake biting its own tail. Kekulé awoke from his reverie and the structure of benzene was solved!
Did Kekulé truly have a sudden prophetic dream solution? It’s impossible to verify someone’s recount of their internal thinking, and some have speculated on this story being embellished in order establish a legacy due to being retold only many years later by Kekulé at the “Benzolfest” in honour of his contributions to chemistry. It was unlikely a true bolt from the blue, as a more dramatic telling to first year chemistry students might go. These dreams often clarify conscious suspicions — organic reactions was Kekulé’s profession and solving structures a waking obsession. A defense of it truly being from a dream has been argued from studying contemporary letters: in his time nobody refuted either Kekulé’s primacy claim on clearly proposing a symmetrical hexagon carbon ring, or his disposition to inspiration from visual daydreams. It certainly didn’t stop Cormac McCarthy from using the story as a launching off point to argue that language is a virus (I find Laurie the most persuasive here).
Of course, a snake grabbing its own tail is not an invention of Kekulé, the Ouroboros is an ancient and world-spanning icon symbolising eternity. Interestingly, the ouroboros has a direct throughline to chemical history through its use as a sygil in alchemy. It expressed the unity of all types of matter, with the ultimate project of course being the profitable demonstration of mastery over matter by gaining the ability to turn lead into gold.

The ouroboros has a long history, including a connection to alchemy where it is illustrated here in relation to chrysopoeia (“gold-making”) through balancing “one in the everything”. —“Chrysopoeia of Cleopatra the Alchemist”, pseudonymous, 3rd or 4th century CE.
The dreamsnake again rears its head (and heads its rear) in the novel Gravity’s Rainbow. The WWII IG Farben chemical colour cartel functions as a constant ominous industrial spectre stalking the book. It tragically mirrors how scientifically and commercially pioneering chemical companies were wrested into the control of war leaders to commit inhuman acts — the ‘Them’ of the military-industrial complex that haunts the book’s entire narrative. Kekulé’s serpentine solution to benzene’s structure is repurposed as a metaphor of the economic boon and destructive cycle unleashed by the WWII war economy:
“Kekulé dreams the Great Serpent holding its own tail in its mouth, the dreaming Serpent which surrounds the World. But the meanness, the cynicism with which this dream is to be used. The Serpent that announces, “The World is a closed thing, cyclical, resonant, eternally-returning,” is to be delivered into a system whose only aim is to violate the Cycle. Taking and not giving back, demanding that “productivity” and “earnings” keep on increasing with time, the System removing from the rest of the World these vast quantities of energy to keep its own tiny desperate fraction showing a profit: and not only most of humanity—most of the World, animal, vegetable, and mineral, is laid waste in the process…
No return, no salvation, no Cycle—that’s not what They, nor Their brilliant employee Kekulé, have taken the Serpent to mean. No: what the Serpent means is—how’s this—that the six carbon atoms of benzene are in fact curled around into a closed ring, just like that snake with its tail in its mouth, GET IT?”
— Gravity’s Rainbow, Thomas Pynchon, pg. 412, Penguin 2nd Ed.
The connection to colour and dyestuffs in the novel does not end there; the essay “Coloring Gravity’s Rainbow” applies a critical lens to these chromatic and chemical connections and charts how they shift throughout the book. The protagonist’s very name —Tyrone— could interpreted to be linked with Tyrian purple, his colour depiction alters as the narrative shifts towards an annihilating white, and the obsessive subjugation of nature through nomenclature and technology is linked explicitly to the terms of the organic chemistry industry.
The connection between the dye industry and the ability to wage war was already well noted in the period between the world wars, as an American industrial pamphlet from the 1920s declares:
“Who makes dyes today can tomorrow make high-explosives”
—“World Disarmament and the Master Key Industry”, American Dyes Institute, 1921.
How ring stability enables destruction and cures 💥💊
The lack of reactivity of benzene was puzzling to chemists ever since it was first isolated. The high C:H ratio entails locations where carbons are double-bonded to themselves, leaving an opening for positions for where atoms could be attached if the double-bond could be broken during an addition reaction. It is now clear that the carbon rings in benzene and benzenoids are unreactive because their bonding arrangements are already stable. However, that does not mean benzene derived compounds are inherently inert. Quite the opposite, the core stability in the centre of benzenoids has led to the creation of molecules that either have phenomenal longevity or massive destructive power depending on what’s attached to the ring. These aromatic molecules have become notorious globally in their own right, with entire departments required to mitigate their danger to human life.
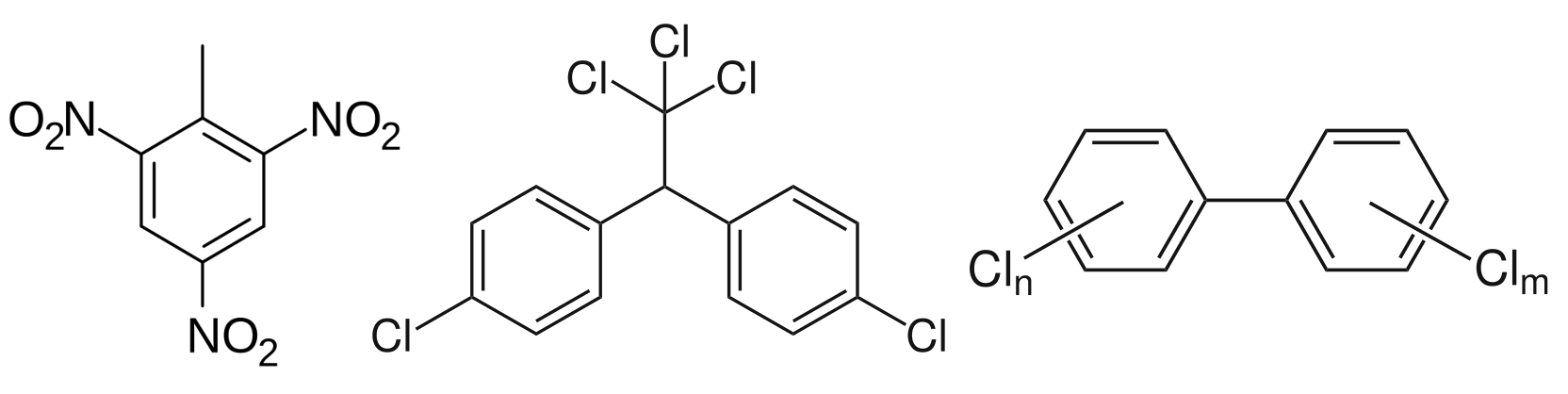
(In)famous aromatic structures that led to: (TNT, left) a profitable explosive and arms company that funded the creation of the Nobel Prize, with Alfred Nobel believing in a 20th century version of mutually assured destruction, earning himself a mistaken obituary claiming that he “could hardly pass as a benefactor for humanity”; (DDT, centre) the founding of the US Environmental Protection Agency under pressure from an exposé on pesticides; (PCB, right) industrially valuable capacitor materials, that nevertheless are carcinogens that have persisted after their global ban by the 2001 Stockholm Convention.
The reason for the proliferation of organics containing these aromatic rings is the central hexagon (or more rarely pentagons and other polygons) supplies a scaffold that provides more lateral directions to build out the bonds of a molecule. Small aromatics are also readily bioavailable, and hence frequently absorbed as toxins or cures (indeed many of your own key biomolecules contain aromatic rings). Ring systems are everywhere in pharmaceuticals. Small organic molecules containing ring scaffolds describes many types of drugs including: opioids, painkillers, psychotropics, antibiotics, anti-inflammatories, etc, etc.
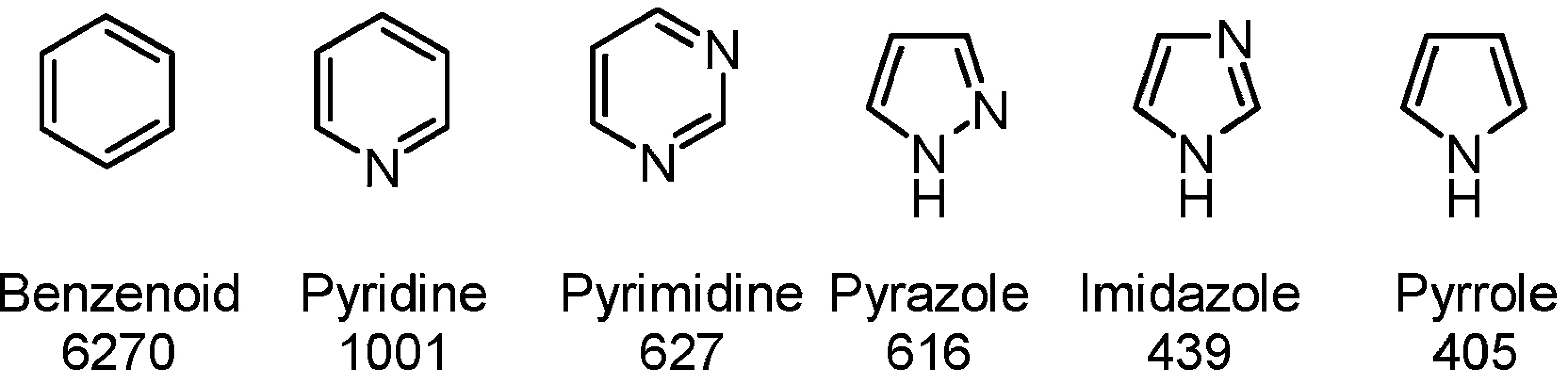
An example of how often different rings occur within a 3566 compound dataset (they can occur multiple times in a structure). — Roughley, S.; Jordan, A., “The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates”, Journal of Medicinal Chemistry, 2011, 54, 10, 3451–3479
The birth of “resonance” and fictitious intermediates ⌬ ↔ ⏣
So, what did Kekulé’s benzene look like? While it’s commonplace to call the drawing of a hexagon with static alternating single and double bond lines the “Kekulé structure” to distinguish it from the “Dewar benzene” that is bifolded by a single-bond through the middle (which can exist, and Dewar also didn’t back the structure that carries his name), these chemists were too empirically-minded to be drawn on a fixed depiction. What is clear instead from the drawings in Kekulé’s papers is an unsatisfied valence in a C\(_{6}\)H\(_{6}\) chain which is only satisfied when the chain is closed by attaching back to its beginning.
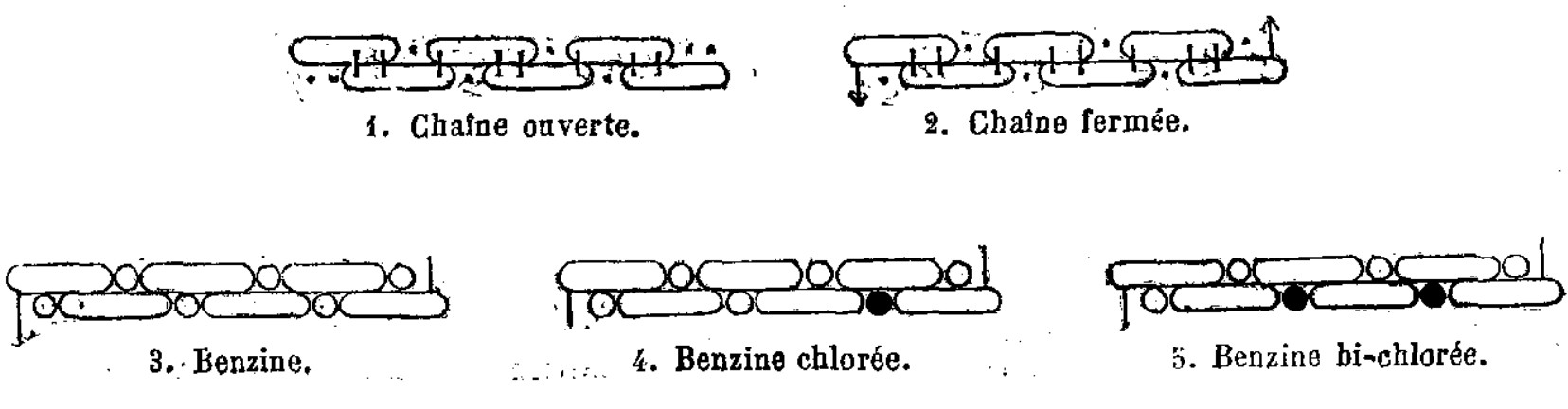
Kekulé’s schematic of benzene illustrates a sense of overlap, but doesn’t accord with current ways of representing organic molecules. —“Sur la constitution des substances aromatiques”, Kekulé, M., Bulletin mensuel de la Société Chimique de Paris, 1865, 98–111.
The oblongs were not meant to show that the molecule literally lay in a straight line, but what the sausage atoms (literally wurst in his articles) were meant to represent was contemporary chemical thought which held the valency property of an element was due to multiple subparts each capable of forming a connection. This is clearer in other diagrams where these particles of ‘atomicity’ are drawn like they’re wrapped in a sock to form the overall element.
{kind=link}
While purely a trick to be able to depict the multibond connections available to these elongated multivalent elements, the dimensional limitation of these sketches seems to have led Kekulé to some mistaken structural assignments, such as initially assigning a triangular form of benzene to achieve chain closure. However, other organic chemists immediately picked up on this based on experimental evidence of 6-fold positional symmetry (additions to the benzene ring creating isomer patterns that are unique to a regular hexagon). Regardless, Kekulé derived the need for complete symmetry in the ring from carbon’s tetrahedral tetravalent bonding. His supposition was not of alternate structures in equilibrium, but that relationship of each carbon to its ring neighbour was held together in equal spacing, mediated by oscillating collisions with two neighbouring carbons and one hydrogen. Kekulé’s daydreamed timescale was likely intramolecular vibrational, not reactive, nor electronic (since the electron was only discovered the year after he died).
The importance of the number of carbon atoms (often six) was recognised by early chemists as well. Armit and Robison popularised the term “aromatic sextet” through many persuasive molecular examples that differed greatly from benzene but still maintain rings of six delocalised electrons. However it was Ernest Crocker, a scientist who never pursued the “Doctor” title, who got there first. He did this by applying the valence electron “octect rule” to all atoms in benzene with Lewis-Langmuir bonding and accounting for the aromatic stability from the remaining six electrons. He places the electrons clearly “outside” the ring due to charge considerations, but states that: “electrons are continually vibrating in the plane of the ring… The net physical effect… serving to bind together all carbon atoms present into virtually a single atom.”
These days, it is common to introduce students to benzene’s cycle through a smearing of two “resonance” structures. We come to a resonance structure due to a dogged adherence to Lewis bonding and “formal charges” as a pedagogical accounting tool for building up (“aufbau”) bonding lines from atoms. Two valid Lewis structures, with alternating double bonds, are drawn with “resonance equilibrium arrow” between them (though the reality being “1.5” bonds is generally acknowledged). So, how did we arrive at two so-called ““Kekulé”” structures of benzene if the early pioneers and experimental evidence evinced a complete symmetry in the nature of benzene? 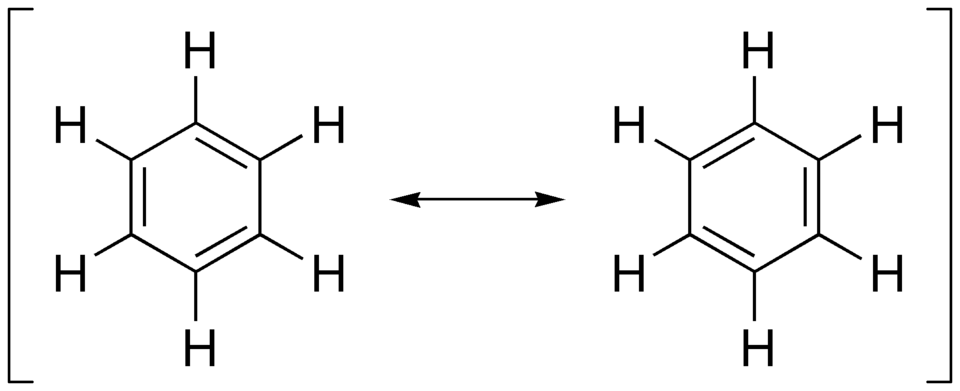
The two “resonance contributors” of benzene, where the single and double bonds are shown to alternate in viable positions.
Likely the instructional ease of drawing Lewis bonding structures helped the misnomer “resonance” stick. I’ve labelled it a misnomer as there is not a “resonation” allowing for two nuclear or bonding position extrema to interconvert in equilibrium; benzene in an unaltered state is a purely symmetrical (\(D_{6h}\)) flat hexagon. What the resonance concept does is bring the concept of energetic degeneracy (i.e. multiple quantum states that have identical value upon measuring a specific observable) into the chemical drawing system learned by practising chemists. The adoption of this useful fiction was turbocharged by populariser extraordinaire: Linus Pauling.
Linus Pauling ran an exposition campaign to bring the insights of the new quantum mechanical theories of bonding to chemistry departments. While Pauling’s explanations focused on chemical conceptual accessibility rather than the rigours of strict physical chemistry research, this approach was widely popular. It was couched in a visual process that unlocked the conceptual basis of structural chemistry in a way that could be latched on to by scientists designing organic synthesis reactions. While Pauling may be responsible for popularising electron delocalisation concepts in language and rules palatable to the synthetic chemists of his day, Fritz Arndt may be the source for the double-headed “resonance equilibrium” arrow that can mislead some chemistry undergraduates. Arndt published papers expounding on a “Zwischenstufe” (intermediate state) in resonance stabilised structures, though to Arndt’s credit he never committed to an oscillating model, or timescale, and emphasised purely electronic bonding rearrangement in a pre-quantum mechanical explanation. Decades later Pauling’s prestige, accumulation of credits, and renowned teaching skills would generate a best seller, popularise a dubious concept, that led to a lot of wasted money and time despite a bongo accompanied ode to orange juice from an equally lionised scientist going through his final days.
The Soviets, however, were having none of it. Resonance structures were decried as “anti-realist” and antithetical to the true teaching of dialectical materialism. The fictitious mathematical idealisation of real atomic positions was viewed with suspicion, and the Soviet chemists adopted a terminological compromise, with the mathematical constructs themselves considered more palatable to a realist philosophy than these bourgeois anti-realist “resonance hybrid” notions. To object to such notions is not surprising, as they clash with the official Marxist-Leninist views of a state that held Engels as an authoritative expert on geometric cosmology.
“A motionless state of matter is therefore one of the most empty and nonsensical of ideas – a “delirious fantasy” of the purest water.”
— Anti-Dühring: Philosophy of Nature. Cosmogony, Physics, Chemistry, Friedrich Engels, 1877, Part I, Section IV.
Resonance joins a pantheon of scientific concepts which at one time or another were suppressed by the Bolshevik state, including: Darwinian evolution; Ernst Mach’s philosophical moves from Netwon’s clockwork universe to one that permitted relativity in motion but also sensational experience; early cybernetic thought (which appears to have been shucked down into systems theory in STEM fields, while cybernetics remains a domain of HASS departments); non-Pavolvian physiology; and interpretations of wave-particle duality.
It should be conceded that, while formally fictitious, the concept of resonance hybridised orbitals played an outstanding explanatory role in revealing bonding behaviour in typical aromatic organic molecules. It has been argued they can still be considered a “correct” representation of aromatic molecules (particularly of charged delocalised tautomers) since dominant resonance contributor representations can be constructed from wavefunction-based molecular orbitals by applying natural bonding orbital analysis.
The electronics of dyes ⚛️
Planar, hexagonal benzene geometry was key to advancing the quantum mechanical explanation of its bonding pattern. The six “remainder” electrons after accounting for co-valent bonding do not lie “outside” the carbons. Instead, contrary to the Crocker proposal, the electrons ‘circulate’ both above and below the ring at once. In fact, in the plane of the ring is the one area the “remainder” carbon electron orbitals cannot be due to a node of density absence caused by the angular momentum of the orbitals. Angular momentum is one of the few properties that an electron can have, being particles otherwise indistinguishable from one another. These remainder electron orbitals are commonly described as hybridised — another concept Pauling intuited and popularised from the mathematical constructs created by the likes of Slater. The outer orbitals of carbon each have a one angular momentum “slice” through them, making it a \(p\)-orbital (from now-nonsensical vestigial spectroscopic notation). In the hybridised view, all 4 carbon valence electron arrange into the tetrahedrally splayed \(sp^{3}\) hybrid orbitals to join with 4 neighbours. The cyclic chains in aromatic systems instead permit for 3 \(sp^{2}\) hybridised carbon covalent bonding orbitals, with the “remainder” \(p\)-orbitals supplying ‘free’ electron density whose adjacent densities can join in what is called a \(\pi\)-bond. Because electrons do display wave-particle duality, these ‘wavicles’ can add their amplitudes because their wavefunctions are in phase. \(\pi\)-bonds provide typical double bonds, but what makes aromatic bonding special is continuous, cyclical, \(\pi\)-bonds tracing a ring an electron can flow around.

Chemical aromaticity arises due to a planar cyclic \(p\)-orbital system that can delocalise to form a π-electron system around a closed ring. —LibreTexts Chemistry 15.9: “What Is the Basis of Hückel’s Rule?”, Tim Soderberg (University of Minnesota, Morris)
Hückel solved the quantum mechanical bonding rules of benzene, even as “resonance” was being bandied around, using two constants: \(\alpha\) – the energy of a \(p\)-orbital, and \(\beta\) – the stabilisation gained by allowing adjacent \(p\)-orbitals to delocalise into a \(\pi\)-orbital. This is because electron position and momentum observables follow the Uncertainty Principle; they multiply together to some (Planck) constant. A highly constrained electron has very low position uncertainty, meaning broad uncertainty in its momentum, allowing a high kinetic energy value. Conversely, the delocalisation stabilisation energy \(\beta\) occurs because the electron density is spread over a large position space in an extended \(\pi\)-orbital, lowering the kinetic energy of the system (\(\nabla^{2}\)).
It turns out six isn’t the sole magic number allowing an “aromatic” ring of delocalised electrons, nor must every atom strictly be carbon (if \(p\)-orbitals are still available after covalent hybridisation). So “aromatic” rings can be created whenever \(\pi\)-orbital electrons can loop back on themselves. This leads to the common rule-of-thumb in chemistry.
Delocalised “aromatic” systems arise in cyclic arrangements of atoms, where \(4n+2\) \(p\)-electrons are “shared” between all atoms in the ring; \(n=1,2,3...\)
In fact the overlapping orbitals can be obtained from unusual geometric configurations, even highly exotic twists like Möbius aromaticity. More commonly, several smaller rings are joined together to make one large delocalised electron system. A famous such aromatic scaffold is the porphyrins: four pentagonal nitrogen-containing heterocycles in conjugated linkage, and so named because of their red-purple hues. Research into the physical basis of “aromaticity” is still a hot field in chemistry, just this year scientists made a massive 18-porphyrin ring that displays current across 242 electrons running through the entire system.
{kind=link}
Understanding the electronic basis of benzene’s bonding explained its unreactivity through aromatic stabilisation. But it also explained something more visible: colour.
Tuning the resonant hue 🎨
The origin of the colour of aromatic dyes can be studied when two aromatic rings are strung together with a conjugated carbon chain. This chain provides the length along which the electron wavefunction can form standing waves. This behaviour is effectively a complicated particle-in-a-box constraint. Consequently, lengthening the conjugated chain lowers the frequency of the electronic wavefunction. But for colour what matters is how this electron wavicle interacts with light. A photon can excite the electronic wavefunction from the highest lying resting state, to next available (‘virtual’) standing wave, provided the energies match. In the process the photon is absorbed, leading to the observation of colour due to a spectral gap in the white light reflected from an object. Longer chains will have a smaller occupied-virtual gap, shifting the absorption to redder light and making the dye appear the complementary colour (bluer). Thus, for calculating the colour of dyes, what matters is the gap between the highest occupied electronic state and the next available lowest energy ‘virtual’ state — the excitation energy.
Seth Olsen (vale) was an Australian theoretical chemist, and like many physical chemists the excitation energies of dyes fell inside his many interests. Relevant here is his work using modern computational modelling with molecular orbital theory to provide predictive explanation of Leslie Brooker’s rules of resonant dye colour absorption discovered in the 1940s. Brooker studied dyes with two aromatic ring systems linked by a simple conjugated carbon chain of different lengths. In the process, Brooker discovered a relationship when he contrasted dyes that were symmetrical (both ends the same), versus asymmetrical dyes. The relationship was this:
Dyes with asymmetric ends absorb at a photon wavelength at the mean value of the two symmetric parents, but deviating by a blueshift. This deviation is related to the different abilities of the end rings to pull electrons from the central chain (their “basicity”). The larger the difference in basicities, the greater the blueshift deviation from the simple average.
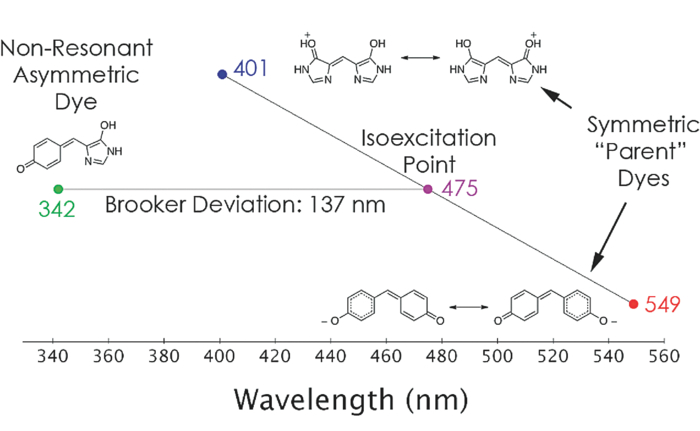
The asymmetrical dye deviates from its “resonant” isoexcitation point — the average of the two symmetric parents. This deviation is directly linked to the electron attractive nature of the end ring systems. — Olsen, S.; McKenzie, R., “Bond alternation, polarizability, and resonance detuning in methine dyes”, The Journal of Chemical Physics, 2011, 134, 114520
Brooker’s Deviation Rule was worked out experimentally within Kodak labs, decades before computational chemistry methods could be used to find explanations. Seth Olsen and Ross McKenzie showed this empirical deviation rule can be explained by molecular orbital theory, though the orbital active spaces involved go well beyond the simple Hückel picture given here, even for classic green and blue dyes. One key result is that the one-electron Brooker basicities can be treated as a constant for each aromatic ring end, regardless of what other ring it is conjugated to, because the localised frontier orbitals remain unchanged. This greatly simplifies the prediction of the excitation energy, meaning the colour of any such dye can be predicted if the constants of each ring system is known — without the need to actually construct them and measure physical values.
By providing a representationally simple two-state model of dyes, these studies yield interesting results for the oxonol dye chromophore housed inside Green Fluorescent Protein (GFP), the most widely used bioluminescent probe in molecular biology experiments. These include that the prevalent form of the dye is the anionic one (since the resonant colour is more easily tuned to fit evolutionary pressures), and the existence of an unobserved ‘dark’ state in GFP absorption.
The molecular orbital picture of dyes can get stranger still. Take azulene, a vivid dark blue isomer of naphthalene. While azulene has been isolated as a dye for hundreds of years by steaming chamomille, research in 2023 reveals that the molecule’s excited state has unique anti-aromatic ring currents which makes it disobey a long standing principle in photochemistry — azulene’s photon emission is not from its lowest lying excited state (Kasha’s rule) but from the 2nd excited state. More widespread but no less entrancing is the class of compounds that are a byword purple itself: the porphyrins. They often function as the molecular holster for a metal atom: the combinations of the transition metal \(d\)-orbitals with the delocalised orbitals extended over the aromatic ring system have beautiful molecular orbital representations, as illustrated below with an iron atom.
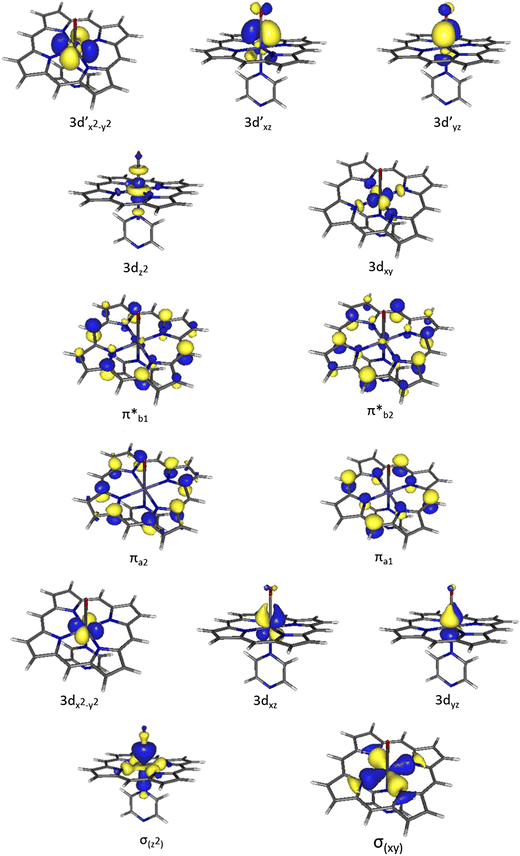
Ten active orbitals in the electronic structure of an iron-porphyrin complex typical of haem proteins. Calculated at the CAS(14e,14o) level of theory with \(A_1\) symmetry. —“Methodological CASPT2 study of the valence excited states of an iron-porphyrin complex”, Journal of Molecular Modelling, 2017, 23, 53.
Porphyrins everywhere!
The porphyrin with greatest recognition is chlorophyll: a modified porphyrin ring holds the central magnesium atom and a poly-terpene hydrocarbon chain extends off as a tail. Chlorophyll is of course the key photon antenna for photosynthesis, and the ultimate input channel of energy into all biological ecosystems, captured from radiation cast off during nuclear fusion. The high school formula 6CO\(_{2}\) + 6H\(_{2}\)O + light \(\rightarrow\) C\(_{6}\)H\(_{12}\)O\(_{6}\) + 6O\(_{2}\) completely hides the spooky resonant action-at-a-distance of absorbing stray photons, channelling their energy to a photosynthetic reaction system, and charging electrons to then drive formation of carbohydrates. Words fail me and I’ll let Drew Berry’s gobsmacking animation of the photosynthesis process for the Walter & Eliza Hall Institute of Medical Research speak for itself.
We started this colour journey with the abundant green from the chlorophyll of plants, but porphyrins are responsible for far greater share of the rainbow displayed by living organisms. Natural colours from porphyrins include:
🌈 Bilins, which are “unwrapped” porphyrins. They display interesting light harvesting properties, e.g. in pychobilin (algae). Their colour spans the rainbow range upon chemical degradation.
🔴 Haem (as in haemoglobin) the source of unmistakable red of oxygenated blood, which is broken down within 3-4 months by your liver to produce…
🟢 Bile, a disconcerting colour to ever see.
🟡 Urine, and other bodily fluids. Unless you are unlucky enough to be suffering from…
🟣 Porphyria disease where badly formed enzymes causes porphyrin build-up, turning your urine purple.
🟤 The brown of egg shells, unless transformed into…
🔵 The blue of a robin’s egg from oocyanin.
⚫ The petrochemical darkness of crude oil and shale, which are full of geoporphyrins, some of the first clues that petrol has biological origin.
🌟 The ‘glow-in-the-dark’ of the Sierra luminous (Motyxia) millipede comes from a porphyrin containing photoprotein whose structure is unresolved (a candidate for AlphaFold prediction?).
.jpg){kind=link}
Retinal: in microbes 🦠 and in our eye 👁️
Life has an even more ancient connection to purple, harking back to rise of the microbes. Bacteriorhodopsin occurs in the “purple membrane” of phototrophic archaea. Similar rhodopsins are found across diverse microbes, including proteobacteria, enabling a protean metabolism — traditional chemical food can be supplemented with a light-driven energy source. The bacteriorhodopsin purple protein operates as a light driven proton pump as animated in this video where the absorption of a photon triggers proton transfers and subsequent isomerisation, causing the protein to change shape.
The structure of bacteriorhodopsin was solved to atomic resolution using cryogenic electron microscopy by Richard Henderson and co-workers in 1990, who would later go on to win a Nobel prize in part due to that work. This partnership of a photosensitive retinal contained inside a rhodopsin protein is found across all domains of light sensing organisms, including the structures solved this year for the Asgard archaea domain of life.
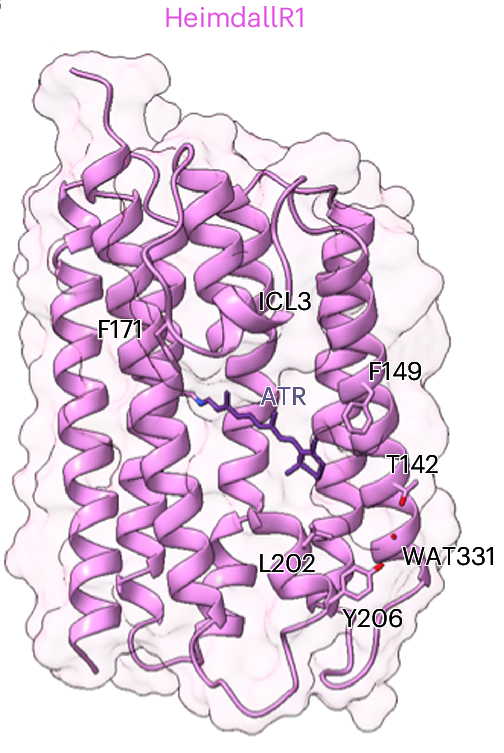
Structures of HeimdallR1 rhodopsin with the retinal chromophore, energy minimised using a QM/MM method. —“Structural insights into light harvesting by antenna-containing rhodopsins in marine Asgard archaea”, Nature Microbiology, 2025, 10, 1484–-1500.
The purple membrane shares a remarkable similarity with vision — not through direct ancestry but through convergent evolution. Microbial proton-pumps (Type I) and visual opsins (Type II) both adapted to use retinal as the light harvester to conduct their work. Type II opsin molecules are directly evolutionarily related to each other, while Type I pumps show strong structural conservation, indicative of shared ancestry even without much sequence similarity. Amazingly, both use variants of the same photochemistry shown above: rotation around the double-bonds upon light absorption.
Which brings us full circle. Purple gave nature rhodopsin, allowing microbes to feed off light. Evolution rediscovered this molecular trick for the opsins that now mediate our vision, (watch “Phototransduction: How we see photons” for the best explanation I’ve seen), while our cones allow us to gaze on purple in all its power and write a post of purple prose about it.
Purple is my favourite colour :)
Deep Purple is shrouded in mystique, while there’s something spectrally special about High Violet hinting at energies just beyond human perception, pushing into the Ultraviolet.
{kind=link}